

Building facilities for pharmaceutical and biotech operations is far from ordinary. Every decision, from material selection to HVAC design, must align with stringent FDA and GMP (Good Manufacturing Practices) standards. Here's what you need to know:

When constructing life sciences facilities, understanding the FDA's key regulations is essential. For most pharmaceutical and biotech projects, the foundation lies in 21 CFR Parts 210 and 211, which outline the minimum GMP standards for drug manufacturing in the U.S.
21 CFR Part 211 delves deeper into facility requirements. For instance, Subpart C (21 CFR 211.42) mandates that buildings be of "suitable size, construction, and location", ensuring they support proper cleaning, maintenance, and contamination control. This includes having dedicated, physically separated spaces for raw material storage, manufacturing, and aseptic processing. Subpart D (21 CFR 211.63) focuses on equipment design, requiring that surfaces in contact with drug products be non-reactive and easy to sanitize.
For digital systems, 21 CFR Part 11 governs the design and validation of building automation and environmental monitoring systems. These systems, which log critical quality data, must meet stringent Part 11 requirements.
| Regulation | Focus Area | Construction Impact |
|---|---|---|
| 21 CFR 211.42 | Facility Design | Requires logical flow, spatial zoning, and defined areas for sterile vs. non-sterile operations |
| 21 CFR 211.46 | Ventilation & Air Filtration | Mandates HVAC systems controlling pressure, microorganisms, dust, and humidity |
| 21 CFR 211.63 | Equipment Design | Requires appropriate size, location, and design for intended use |
| 21 CFR 211.67 | Equipment Maintenance | Mandates written cleaning and sanitization procedures |
| 21 CFR Part 11 | Electronic Records | Requires validation of computer systems used for monitoring and data storage |
FDA inspections for GxP compliance occur at least once every two years [6]. Interestingly, the top issue flagged in 2022 was the lack of written procedures or failure to follow them [6]. This highlights how documentation errors, often rooted in the construction phase, can lead to compliance problems.
These regulations ensure that GMP principles are integrated into every phase of facility construction.
GMP principles influence every decision during construction, shaping everything from material choices to room layouts. At its core, GMP focuses on contamination control. Facilities must follow a unidirectional flow - from raw material intake to finished product shipping - ensuring clean and dirty materials never cross paths. This requires planning for airlocks, gowning rooms, and pressure cascades right from the design phase, rather than adding them later. For example, ISO 14644 specifies that an ISO Class 5 cleanroom used for aseptic filling must have 240–360 air changes per hour using HEPA filtration [7].
Material selection is equally critical. Floors, walls, and ceilings must be seamless, non-porous, and resistant to harsh cleaning agents. 316L stainless steel is the gold standard for surfaces that come into contact with products, while epoxy-coated panels and coved vinyl are common choices for walls and floors. These materials are designed to endure constant sanitization without degradation.
"The 'c' means 'current' - your methods need to keep up with new technology and science." - Jessica R., GMP Pros [5]
Validation is another key aspect of GMP construction, achieved through the IQ/OQ/PQ lifecycle:
This validation process must be integrated from the start, not treated as an afterthought once construction is complete.
Incorporating compliance into the early planning stages is essential, and a risk-based approach can help mitigate potential issues. The FDA uses a risk-based oversight model, meaning higher-risk operations face greater scrutiny. Construction teams that understand this approach can proactively address vulnerabilities during planning.
Tools like Failure Mode and Effects Analysis (FMEA) and Hazard Analysis and Critical Control Points (HACCP) are invaluable for identifying potential risks. For example, applying FMEA to an HVAC system might reveal that a failure in a pressure differential sensor could compromise an ISO 7 cleanroom. This insight could lead to designing redundant sensors into the system, avoiding costly fixes later. Such proactive risk analysis aligns with earlier insights on preventing expensive design changes.
Staffing decisions also play a role. Embedding quality assurance (QA) and regulatory specialists during the pre-design phase - rather than waiting until construction is complete - allows teams to identify and resolve risks early, saving time and money.
"cGMP compliance is required by law. The FDA checks if you follow the rules through surprise inspections at your facility." - GMP Pros [5]
Unlike ISO certifications, which are voluntary, cGMP compliance is legally mandated [5][6]. A single compliance failure during construction could result in an FDA Official Action Indicated (OAI) classification, potentially halting operations worth billions of dollars [7]. This underscores the importance of risk-based planning to safeguard the investment.
A compliant life sciences facility begins with a well-crafted User Requirements Specification (URS). Think of the URS as the blueprint that guides every decision - whether it's about materials, room layouts, or system specifications. Without this foundation, projects can spiral into unnecessary complexity in some areas while leaving critical compliance gaps in others, often leading to costly overruns [4].
To get it right, involve process engineers, QA teams, and regulatory experts early on. This collaboration ensures that compliance shapes the design from the start, making certification a smoother process rather than a stressful hurdle.
One of the first and most critical decisions in this stage is planning the flow of personnel and materials. Facilities must maintain strict, unidirectional movement - from receiving raw materials to final shipping. This approach ensures segregation and cleanliness, with specific zones designed for aseptic filling (ISO Class 5) and others for support activities (ISO Class 7–8).
Once the URS is solid, the focus naturally shifts to designing and planning critical systems and utilities that meet GMP requirements.
HVAC systems are the backbone of any GMP facility, demanding precise control over pressure, temperature, and humidity. These systems must maintain pressure differentials of 12.5–25 Pa between rooms, temperatures between 18°C–24°C (64°F–75°F), and relative humidity levels of 30–60% [7]. It's worth noting that pharmaceutical cleanrooms consume energy at a rate 15 times higher than standard commercial buildings, with HVAC systems alone accounting for over half of the total electricity use [7].
To manage the complexity of utility systems, early involvement of contractors can help align timelines and reduce risks, especially when integrating these systems into the broader project delivery framework.
Clean utilities like purified water, clean steam, and process gases also require meticulous planning. For example, piping must be sloped for complete drainability, and "dead legs" - sections of pipe where fluids can stagnate - must be eliminated to prevent microbial contamination. These design elements are critical for FDA compliance and are often scrutinized during inspections. Steps like video inspections of welds and physical verification of slopes before system closure are non-negotiable [1].
Once the design and utility plans are in place, the construction phase must prioritize compliance through rigorous documentation and strict change control processes. Documentation is as critical as the construction itself. Weld logs, material certifications, and as-built drawings should be maintained consistently throughout the project, not just compiled at the end. Treat the Turnover Package (TOP) as a formal deliverable, with documentation milestones reviewed throughout construction rather than waiting until the handover [1].
"In regulated environments, documentation is as important as the systems themselves. If it's not documented, it didn't happen." - C1S Group [1]
Surface integrity is another area where construction directly impacts compliance. Seamless, non-porous materials like epoxy coatings, monolithic wall panels, and coved corners are essential to eliminate contamination traps and support stringent cleaning protocols [7][2].
Any design changes during construction must go through a formal change control process with documented approvals before implementation [7]. Even minor, unapproved changes can lead to compliance issues during FDA inspections.
Finally, Building Management Systems (BMS) and Environmental Monitoring Systems (EMS) should be integrated during construction, not added later. This ensures that critical data - like temperature, humidity, and pressure - are captured and logged from the moment systems go live [2][8].
Validation ensures that a facility and its systems consistently perform as intended. It’s a key requirement under 21 CFR Parts 210 and 211, essential for FDA approval and successful inspections.
The process unfolds in four phases, each addressing specific questions and activities:
| Validation Phase | Core Question | Activities |
|---|---|---|
| Design Qualification (DQ) | Does the design meet requirements? | Reviewing blueprints against URS and CGMP; confirming material compatibility |
| Installation Qualification (IQ) | Was it installed correctly? | Checking power connections, HEPA filter installation, and verifying serial numbers |
| Operational Qualification (OQ) | Does it operate as intended? | Testing HVAC pressure differentials, alarm settings, and equipment operating ranges |
| Performance Qualification (PQ) | Does the process work under real conditions? | Running systems with actual materials to ensure consistent output quality |
Each phase relies on precise documentation, forming part of the Turnover Package to ensure traceability and readiness for audits.
Avoid vague criteria in validation protocols. For example, instead of saying, "install per manufacturer specs", use measurable benchmarks like "centrifuge rotor speed reaches 15,000 rpm ± 100 rpm." During OQ, include failure simulations to confirm safety features and error detection work as designed. A traceability matrix - linking every requirement from the DQ and URS to specific OQ and PQ tests - can save significant time during an FDA review.
"IQ, OQ, PQ protocols exist to prove one thing: that the equipment you're using will consistently produce products meeting quality requirements. Not occasionally. Not under ideal conditions. Consistently." - The FDA Group [10]
For construction teams, this means embedding monitoring systems during the build phase rather than adding them as an afterthought.
A Turnover Package (TOP) is the formal record handed off from the construction team to the facility owner. It proves that all systems were built, installed, and verified properly. In FDA-regulated facilities, an incomplete TOP could delay commissioning or even trigger regulatory action.
A complete TOP should contain:
The documentation responsibilities can vary depending on the project delivery model, such as design-build, CM-at-risk, or traditional approaches. Understanding these differences is critical to ensuring accountability.
For electronic records, 21 CFR Part 11 requires them to be timestamped, tamper-proof, and accessible only to authorized personnel. Validation must confirm these controls before the facility becomes operational.
"A single compliance failure during construction can trigger FDA Official Action Indicated (OAI) classifications, potentially shutting down billion-dollar manufacturing operations." - CIC Construction Group [7]
The best practice is to treat the TOP as a contractual deliverable, with documentation reviews scheduled throughout the project. This approach avoids last-minute chaos and naturally supports FDA inspection readiness.
FDA inspections for domestic facilities occur at least every two years, though high-risk sites may face more frequent visits based on compliance history and data trends [11]. Solid validation and thorough turnover documentation form the backbone of a successful inspection. The difference between a smooth process and a problematic one often lies in preparation.
Assign clear roles for the inspection:
Set up two spaces: one for the inspector and a separate war room where your team can prepare documents and staff before interviews. Train employees to answer only the specific questions asked, and encourage responses like, "I don’t know, but I can find the answer", instead of guessing [11].
Mock inspections are invaluable. Conduct scenario-based interviews, physical walk-throughs, and checks of every label, door, and visible surface. Remove personal notes and outdated notices. Since data integrity is cited in over 60% of FDA Form 483 observations in 2026 [12], ensure that all records follow ALCOA++ principles - Attributable, Legible, Contemporaneous, Original, and Accurate. Facilities with unified CAPA documentation encounter fewer repeat observations compared to those without centralized systems [12].
"Inspection readiness isn't just about passing audits - it's about fostering operational excellence." - Craig Bradley, SEO Editor, Lab Manager [13]
The goal is to maintain a continuous state of readiness. When compliance becomes part of routine operations, FDA inspections simply confirm what’s already in place.

Life Sciences Construction: Project Delivery Models Compared
Even the most detailed design won't succeed without the right team to bring it to life. Life sciences construction requires professionals who not only understand construction but also have a solid grasp of regulatory demands. The difference between a general contractor and a team experienced in GMP (Good Manufacturing Practices) can determine whether your project passes an FDA inspection or faces costly delays. In this field, assembling the right team is as crucial as the design itself - it bridges strategic planning with on-the-ground execution.
Every regulated facility project hinges on a well-coordinated team of specialists. To understand how construction project delivery works in this space, it’s essential to know who should be involved from the start.
An example of this approach in action is Consigli’s delivery of the Shire Building 400 in Lexington, MA. This 200,000-square-foot FDA-regulated facility earned an ISPE Facility of the Year Honorable Mention. It included the first 2,000-liter single-use sterile train at a commercial scale - an achievement made possible by a highly specialized team [16].
While technical expertise is crucial, the behaviors and habits of a GMP-savvy team can make or break a project. The best professionals combine regulatory knowledge, such as familiarity with 21 CFR Parts 210, 211, and 820, with hands-on experience across the Commissioning, Qualification, and Validation (CQV) lifecycle [9][17]. Strong documentation practices are also a must, as they underpin the validation and turnover processes required for compliance.
In addition to the basics, teams are increasingly expected to embrace Computational Fluid Dynamics (CFD) modeling to optimize cleanroom airflow. Studies show validated CFD use can cut HVAC energy consumption by 10% to 25% [9]. Teams must also excel in Change Control and CAPA (Corrective and Preventive Action) procedures to manage deviations during the project - unmanaged changes can lead to regulatory issues later [18].
"One of the biggest challenges in pharma projects is anticipating compliance and operational needs before the first brick is laid." - Subhendu Mohanty, Vice President, Projects, Pharma Access [9]
The early involvement of process engineers and certification specialists often separates successful FDA inspections from projects that face hurdles down the line [1].
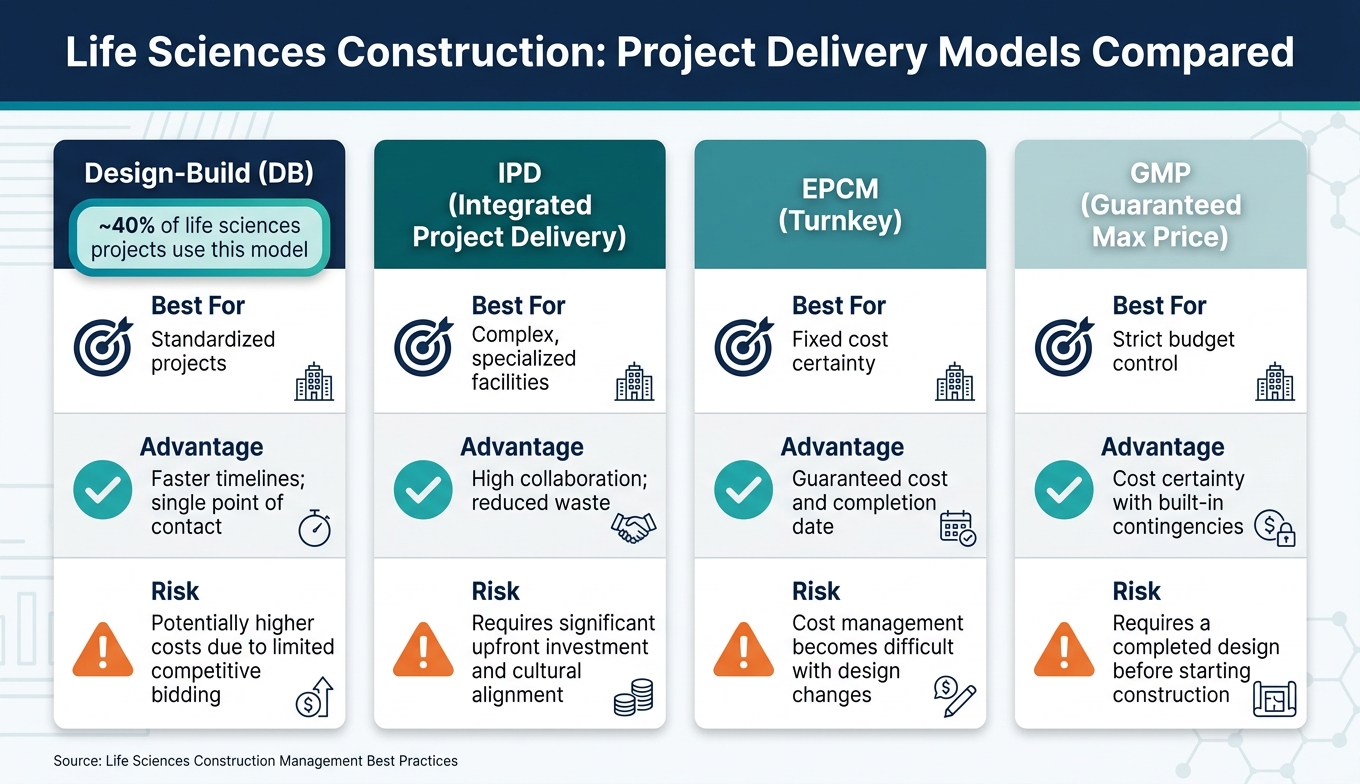
The choice of project delivery model has a direct impact on maintaining compliance throughout the construction process. Each model comes with its own strengths and challenges:
| Model | Best For | Advantage | Risk |
|---|---|---|---|
| Design-Build (DB) | Standardized projects | Faster timelines; single point of contact | Potentially higher costs due to limited competitive bidding |
| IPD (Integrated Project Delivery) | Complex, specialized facilities | High collaboration; reduced waste | Requires significant upfront investment and cultural alignment |
| EPCM (Turnkey) | Fixed cost certainty | Guaranteed cost and completion date | Cost management becomes difficult with design changes |
| GMP (Guaranteed Max Price) | Strict budget control | Cost certainty with built-in contingencies | Requires a completed design before starting construction |
Design-Build is used in about 40% of life sciences projects [15]. Its appeal lies in simplifying accountability and speeding up timelines. However, if subcontractors lack the necessary engineering or BIM skills, design issues can emerge later - when they’re costliest to fix.
Integrated Project Delivery (IPD) is gaining popularity for high-stakes projects due to its collaborative approach. It aligns all stakeholders - owners, contractors, and vendors - around shared risks and rewards. However, it demands a strong cultural fit and higher initial investments. For organizations that can move quickly, EPCM offers fixed costs and schedules but requires partners with local expertise and proven subcontractor networks [15].
"The most common mistake that is seen in the industry is that the subcontractors are left to finalize design instead of the A/E companies." - Gul Dusi, Founder/Managing Director, Dusi Advisory Group [15]
Ultimately, the choice of delivery model depends on an organization’s culture and the complexity of the project. Regardless of the approach, involving project managers early, ensuring clear documentation, and embedding regulatory expertise from the start are critical to successful execution. This integrated team approach lays the groundwork for smoother operations in later stages.
Building facilities for the life sciences industry requires compliance to be deeply embedded into every phase of the project. From cleanroom layouts and HVAC systems to material choices and weld documentation, every decision should align with a clearly defined User Requirement Specification (URS). The workforce and project delivery models you select play a critical role in either strengthening or weakening this foundation.
Take, for example, a $280 million mRNA vaccine facility expansion. By incorporating Facility Risk Assessments (FRA) and System Risk Assessments (SRA) during the early design phase, the team managed to achieve process performance qualification (PPQ) readiness within 12 months. Even more impressively, they cut capital expenditure by 15% in the process [3].
Documentation isn’t just a box to check at the end of the project - it’s a continuous, essential task. Tracking turnover packages, weld logs, material certifications, and as-built drawings throughout the project timeline helps catch potential compliance issues early. This proactive approach to documentation ties directly into regulatory risk management, reducing the likelihood of surprises during inspections.
The biopharmaceutical cleanroom construction market is forecasted to hit $27.80 billion by 2034 [7]. This growth is fueled by significant domestic investments, such as Novo Nordisk's $4.1 billion facility in Clayton, North Carolina, the largest single life sciences investment in the state's history [7]. With stakes this high, every decision - whether related to facility validation or pharmaceutical-specific budget contingencies of 15% to 25% - must prioritize compliance [7].
The projects that succeed are those where compliance isn’t an afterthought but a driving force behind design. Engaging the right specialists early in the process ensures that regulatory standards are met and risks are minimized. As CIC Construction Group aptly points out:
"A single compliance failure during construction can trigger FDA Official Action Indicated (OAI) classifications, potentially shutting down billion-dollar manufacturing operations." [7]
This methodical, compliance-first approach is the surest way to deliver life sciences facilities that meet the industry's rigorous demands.
GMP and QA teams play a crucial role in the facility design process and should be brought in as early as the initial planning and conceptual phases. Their early involvement is key to ensuring the facility complies with regulations, operates efficiently, and addresses potential risks effectively right from the start.
An FDA-ready turnover package generally contains detailed documentation to demonstrate compliance with CGMP (Current Good Manufacturing Practice) regulations. Here's what it might include:
These records are crucial for showing that the manufacturing facility adheres to FDA standards and is prepared for regulatory assessment.
To reduce regulatory risks during construction changes, adopt a clear and structured change management process. This approach helps you assess, document, and approve any modifications before they are implemented, ensuring alignment with FDA, GMP, and ISO standards. Involve regulatory affairs teams early in the process and maintain open communication across all departments to ensure changes meet current regulations. Regularly review and validate these changes, and keep detailed records to prevent non-compliance or regulatory setbacks.








